Hello,
I am trying to solve the structure of a protein complex between a 75kDa protein and a 35kDa protein. I am still unsure about the stoichiometry of this complex, as some of my SEC-MALS-SAXS suggests it is not 1:1, but the particles on my EM grid look like it could only be a 1:1 interaction.
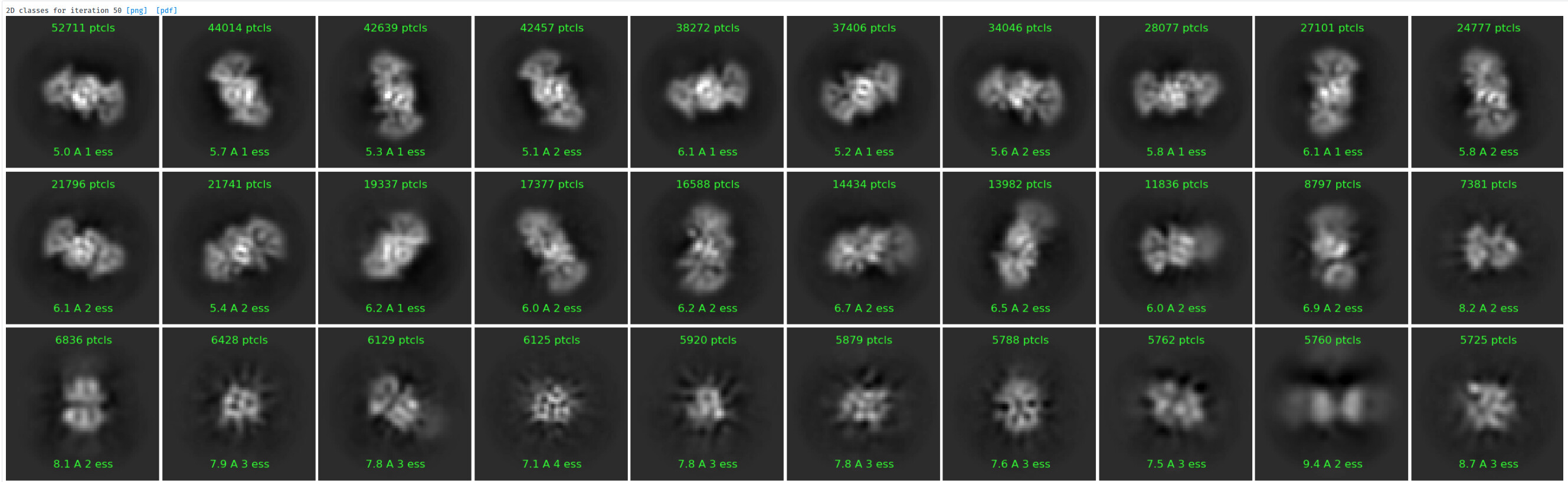
Under the assumption that my complex is indeed 1:1, these are the class averages from my data acquired on a Titan Krios 300keV scope:
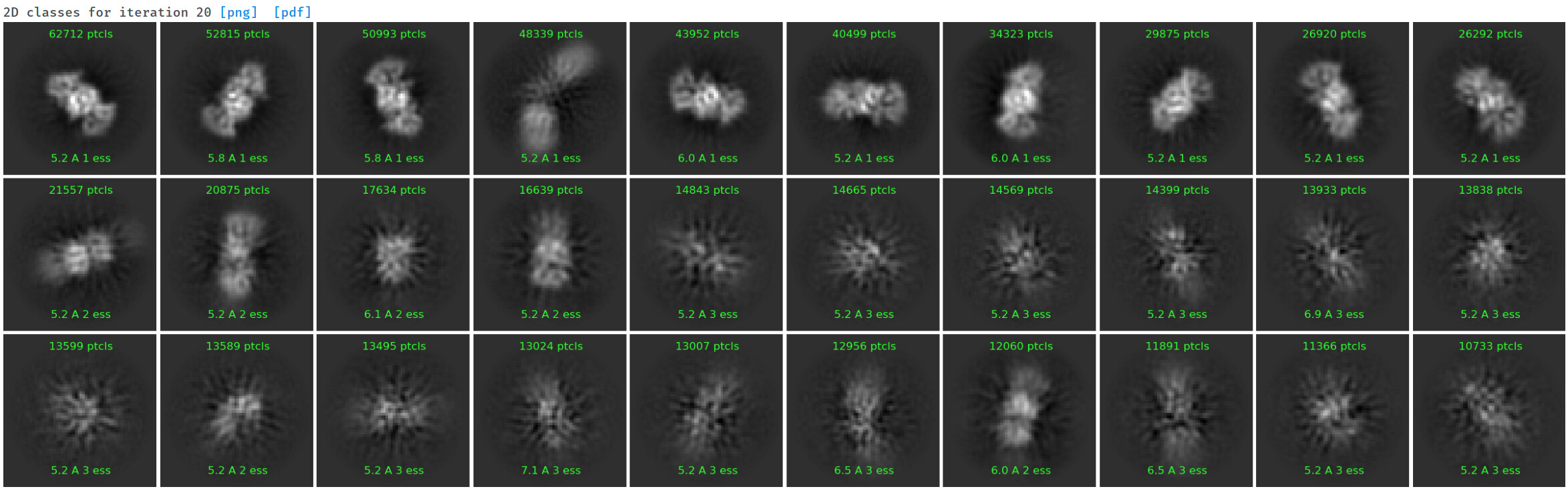
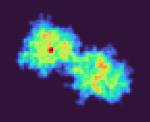
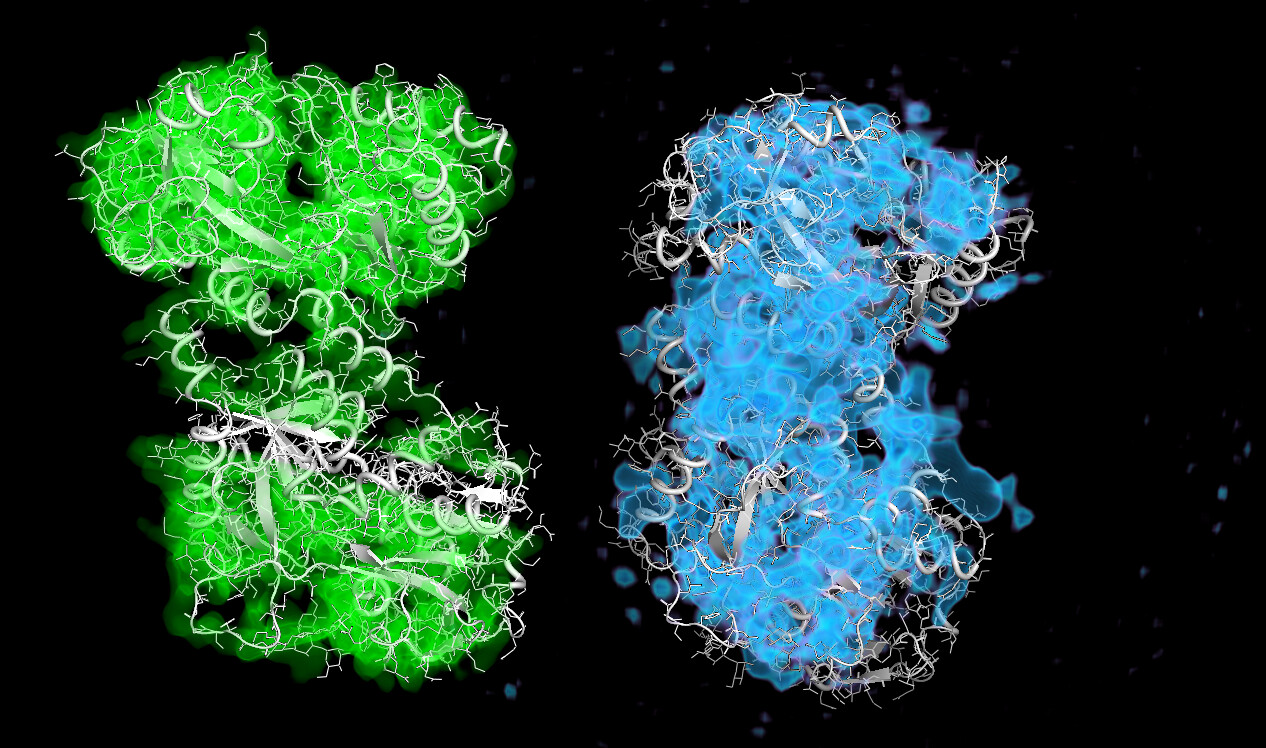
These particles look a lot like my 75kDa protein, apart from the fact that the center of the particle looks like it scatters strongly (higher contrast). If you look at the crystal structure of the 75kDa protein below, you’ll see that the region of this protein with the fewest atoms/electrons is in the center:
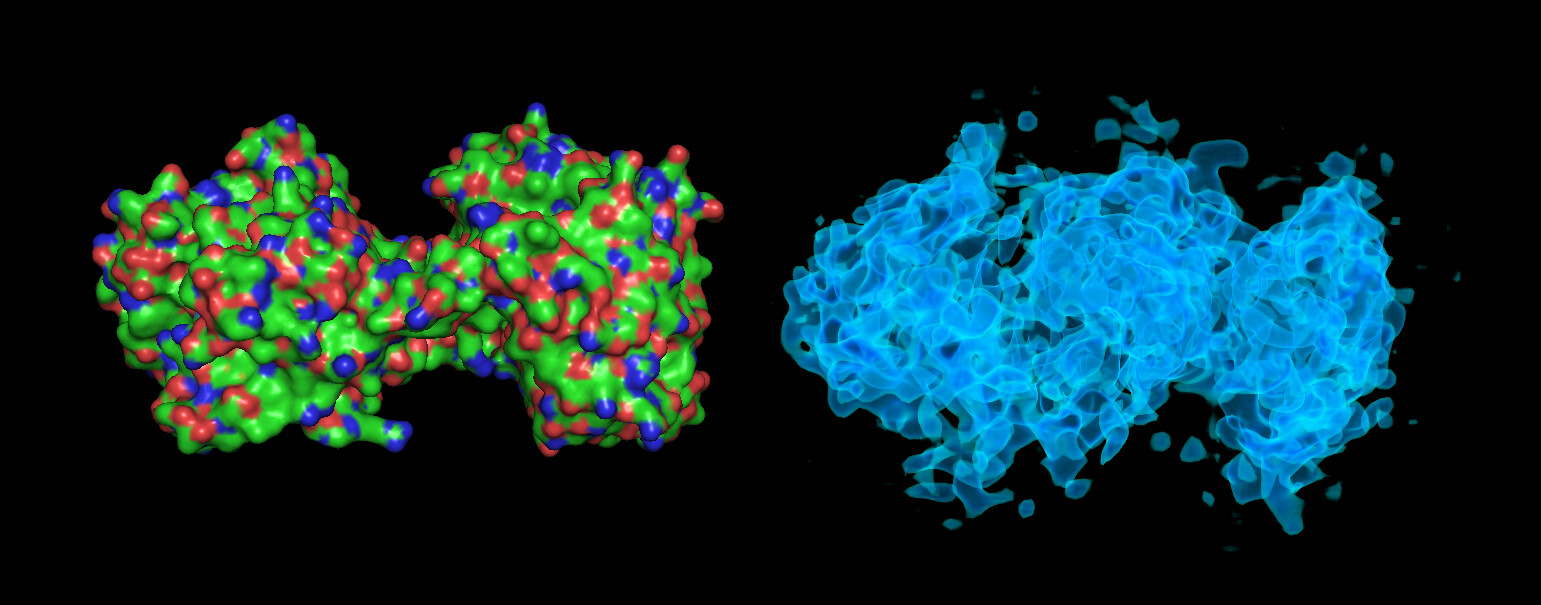
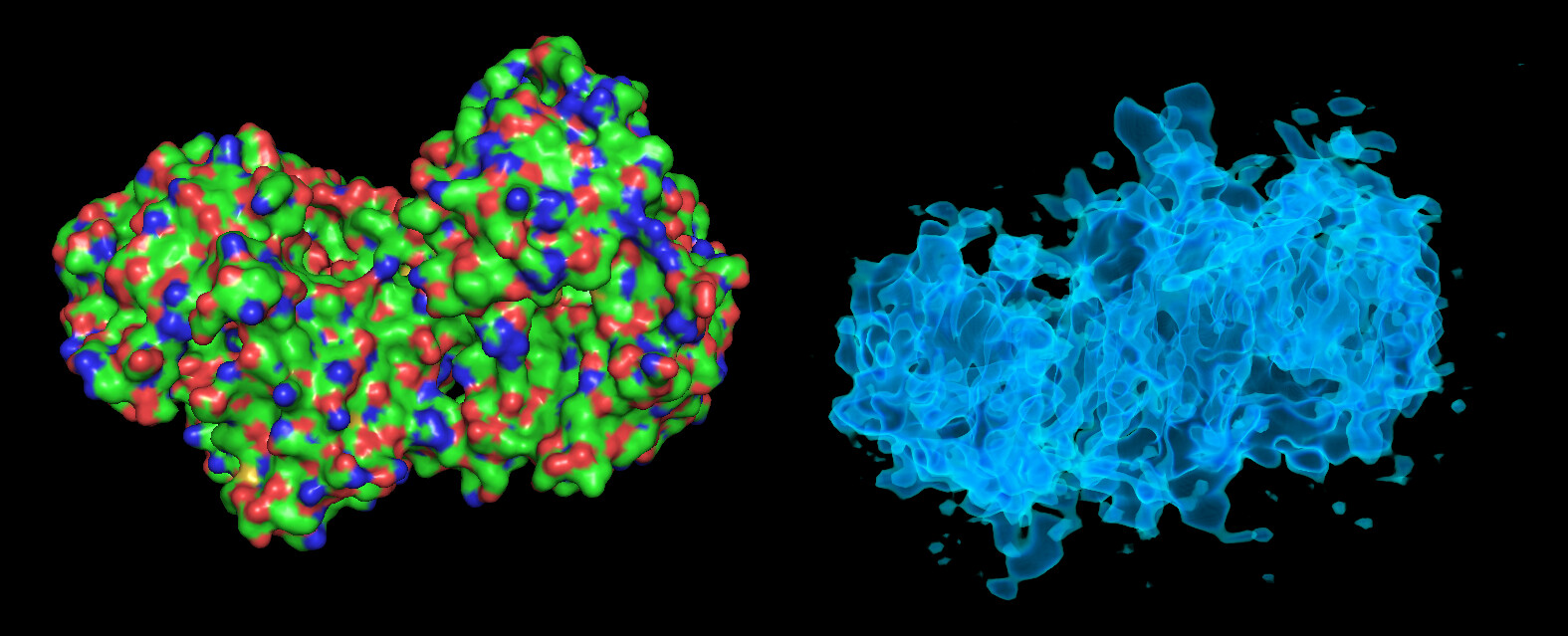
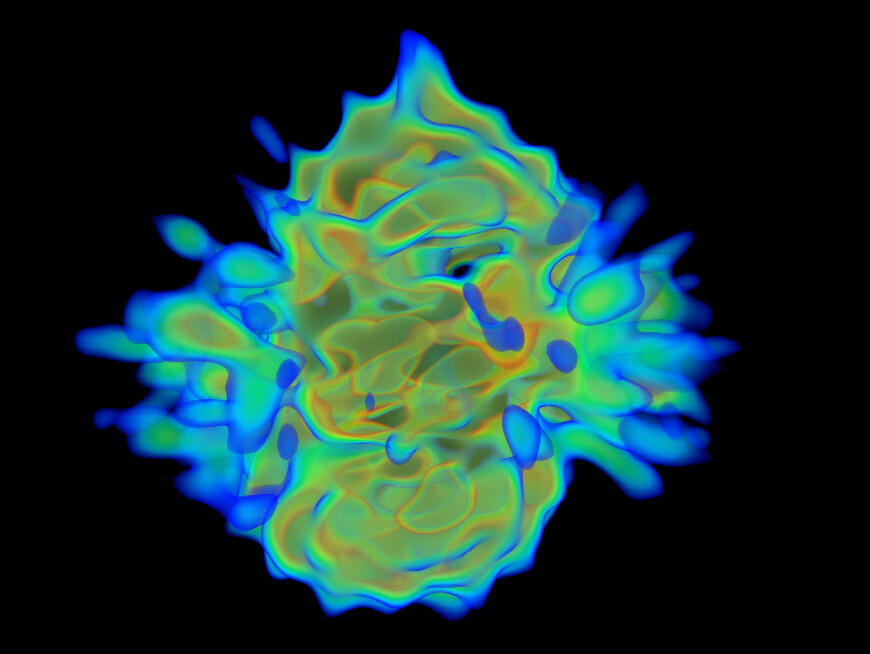
On the right is the map_sharp from my homogeneous refinement. I see, I THINK, density in the middle in the map that doesn’t exist in the PDB. I am hoping this is my 35kDa protein ‘wrapped’ around the center, but I can’t seem to resolve this map to higher resolution to start to see features in that density (I think because I am limited in the viewing angles of my particles… most seem to be from top-down).
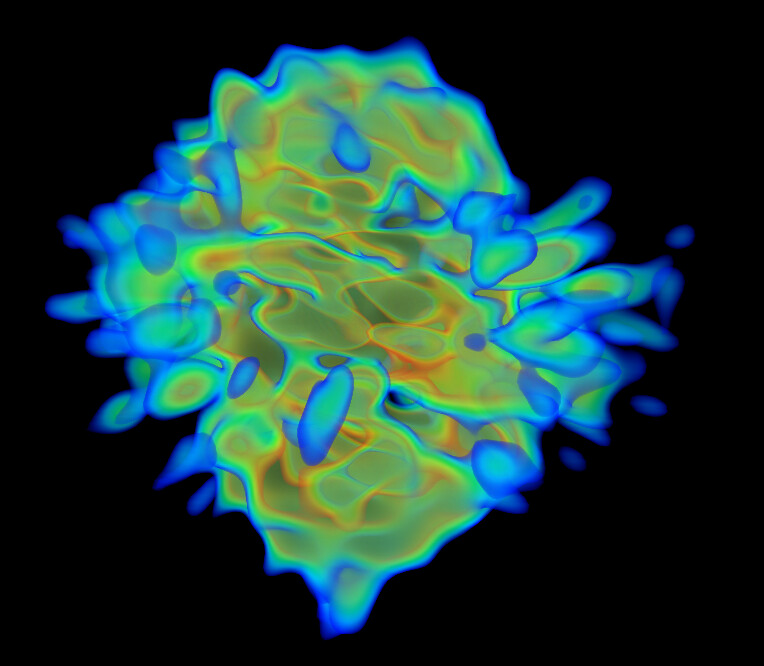
I wrote a script quickly yesterday that will take a PDB and voxelize it, then collapse the voxelized cube into a 2D projection to see what images of this protein should look like if it was on a grid whilst not in complex, and I don’t see any density in the middle, as suspected:
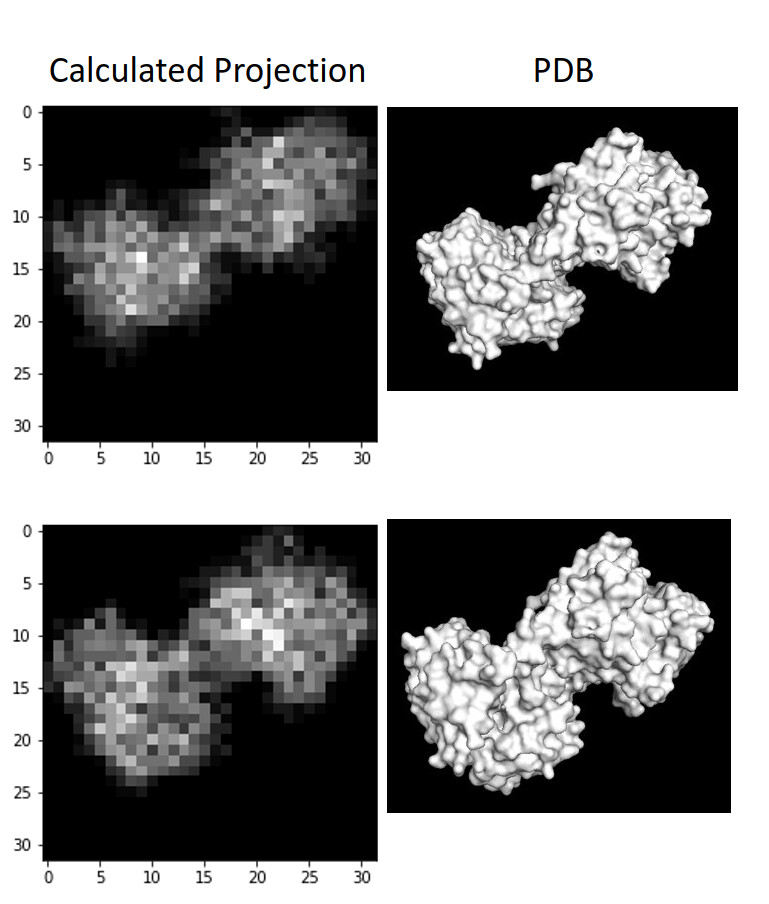
So:
- Is it reasonable to suspect my partner is binding in the center and this map is worth pursuing futher?
- What steps can I take to improve the resolution of my map to start seeing features in this central density?
- Could it be advantageous to acquire a tilt series on this grid to help improve resolution?
I am new to CryoEM, sorry for my noobishness… any help is much appreciated.